Local area network (LAN). Computer and Network Examples
 diagram")
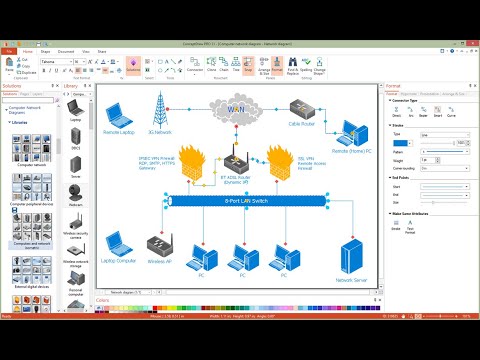
Metropolitan area networks (MAN). Computer and Network Examples
. Computer and Network Examples")
Campus Area Networks (CAN). Computer and Network Examples
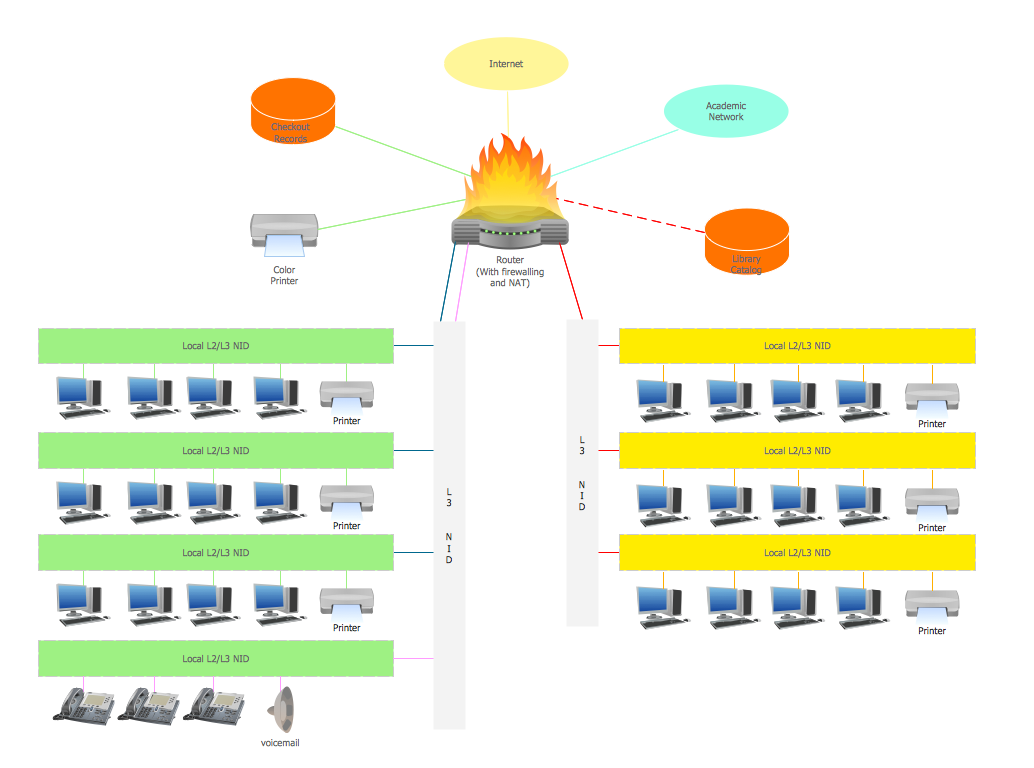
Network Topologies

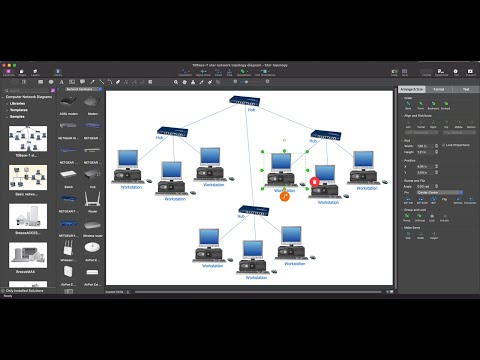
Network Diagram Software Enterprise Private Network
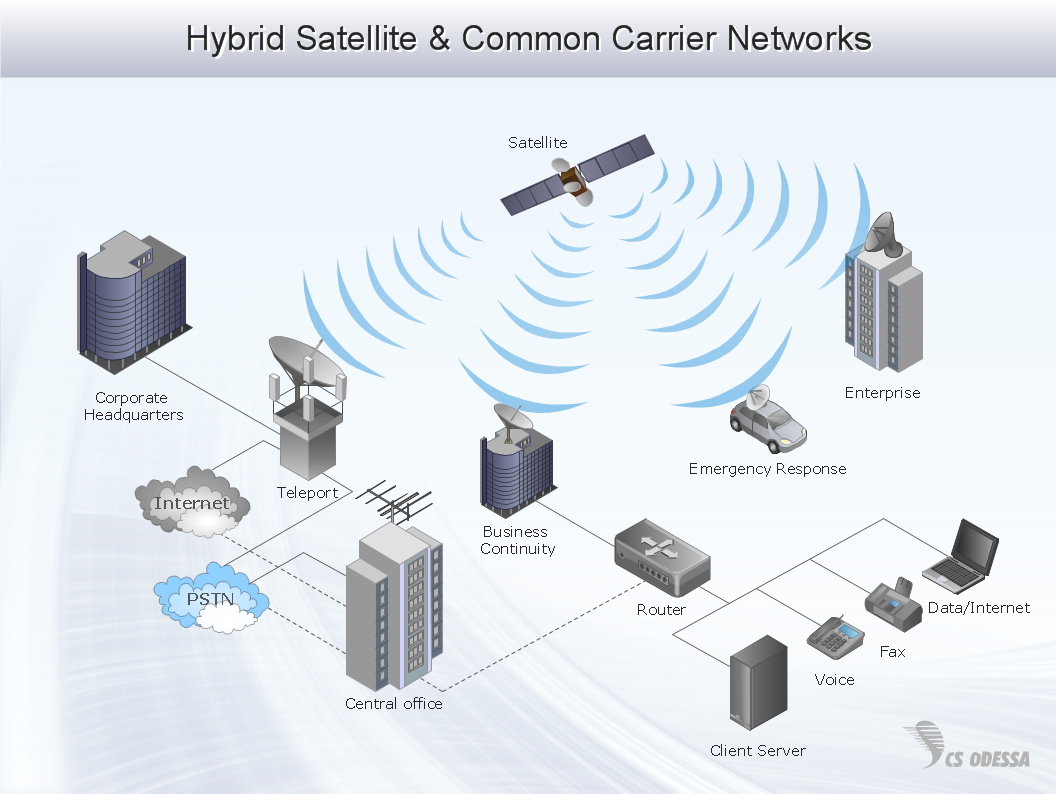
Wide area network (WAN) topology. Computer and Network Examples
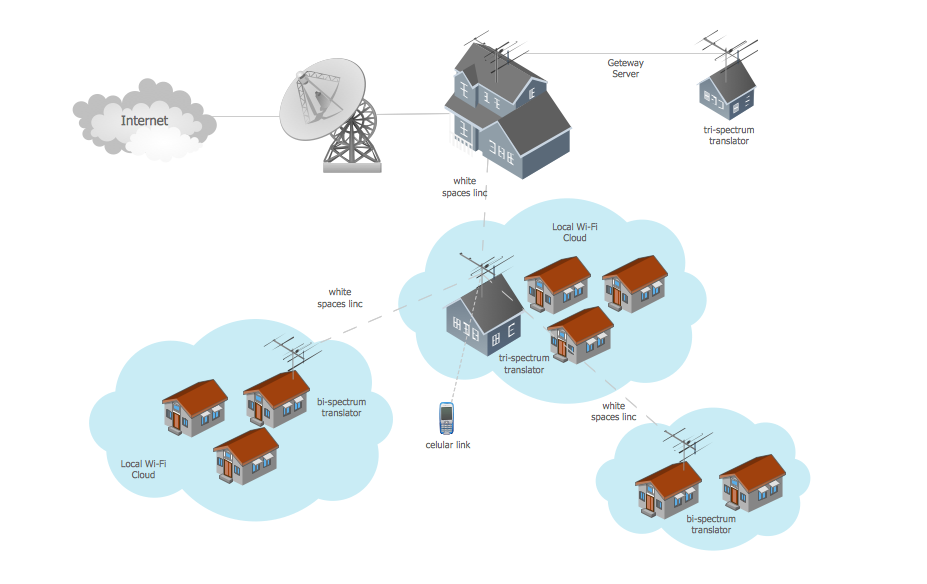
Digital Communications Network. Computer and Network Examples

Storage area networks (SAN). Computer and Network Examples
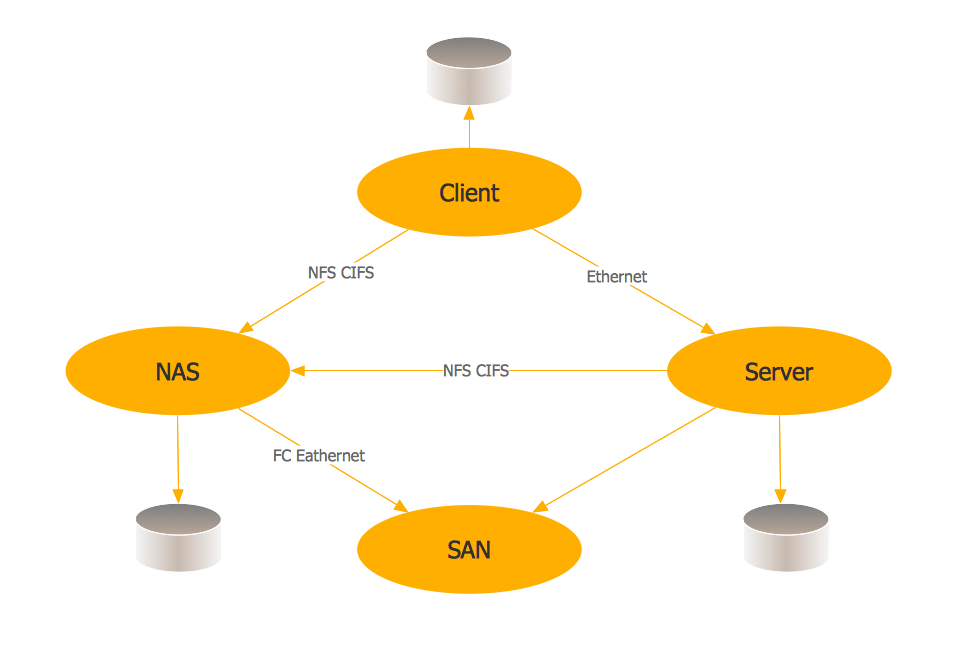
Network wiring cable. Computer and Network Examples
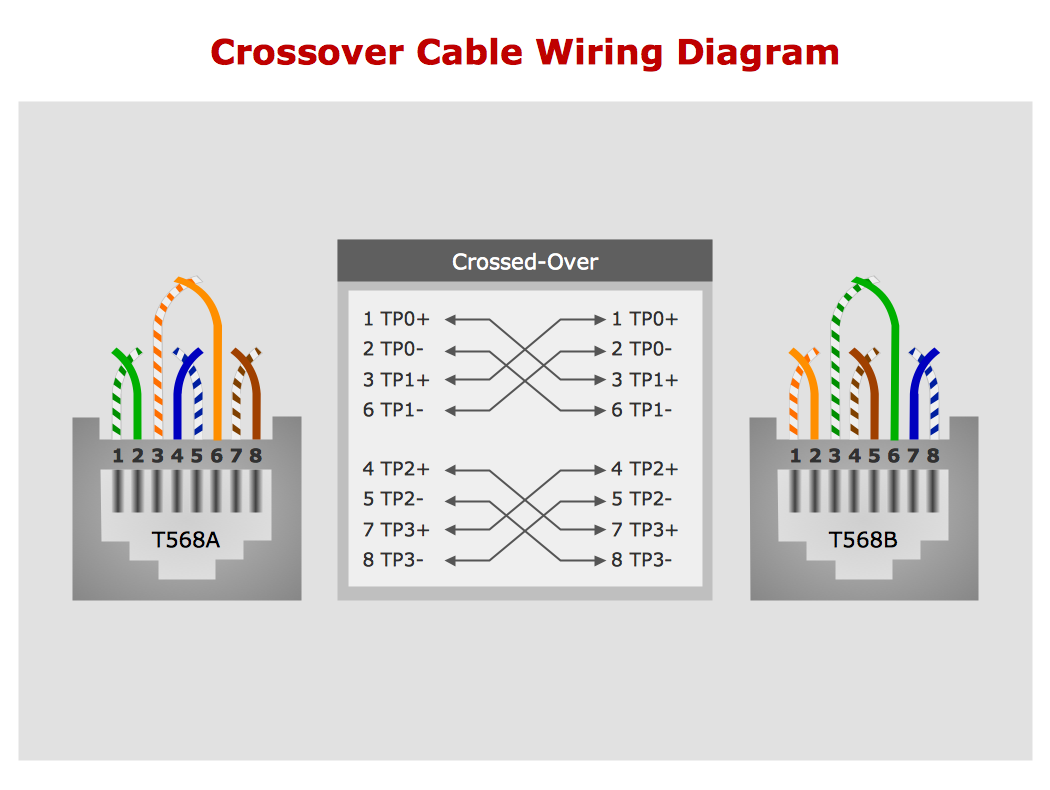
Computer Network. Computer and Network Examples

- Lan Wan Man Network Images
- Images Of Metropolitan Area Network
- Network Pic Lan Wan Man
- Images Of Lan Man Wan Pan
- Lan Wan Network Image Download Hd
- Metropolitan Area Network Images
- Pictures Of Lan Wan Man In Computer
- Types Of Network Lan Man Wan Images Download
- Computer Network Diagrams | Types Of Network Picture Lan Wan Man